Industry Benchmark Dataset and More from QCArchive now Available on Zenodo
Written by: Jennifer Clark, PhD
Through Innovations with QCArchive, OpenFF can Host Datasets on Zenodo
OpenFF data is always freely available on QCArchive, a quantum chemical (QC) data management platform providing a server for data access and storage and software for interaction and management. We are grateful for the Molecular Sciences Software Institute (MolSSI) for this service, as we rely on them daily. A recent development of QCArchive offers the ability to create and download datasets locally in the SQLite format with compressed contents, referred to as “dataset views”. Leveraging this new feature, our key datasets for fitting and benchmarking force fields are now archived and available on Zenodo.
Available Datasets Include Our Popular Industry Benchmark Dataset
Our flagship datasets are available on our website. Notably, our Industry Benchmark dataset [1], which we utilize to benchmark our own research [2-5], has garnered significant attention in external publications [6-10] and online content, including blog posts [11] and code bases [12,13]. Consequently, we have released a new, filtered version of the dataset, Industry Benchmarking v1.2, which eliminates unphysical ring structures. Please continue reading for a comprehensive exploration of the dataset’s contents.
How Accessible is a QC Dataset on Zenodo?
This new offering of datasets on Zenodo aims to provide redundant and enduring accessibility. Zenodo is an open digital archive established by CERN, offering a stable storage option. This choice of SQLite, the standard QCArchive dataset view format, provides our data in a format that is ubiquitous and independent of niche tools. Within these SQLite datasets are the details of quantum chemical calculation specifications and results, with molecular data serialized with msgpack and compressed with zstandard, a combination that provides data with a lossless reduced size. In addition to the common SQLite readers, these datasets can be imported into QCArchive datasets using the following code:
dataset = qcportal.load_dataset_view(filename)
To ensure timeless accessibility, we also provide a Docker image capable of processing these datasets that contains the current Python and QCArchive versions and their dependencies. Upon spinning up a Docker container, a user will be met with a Jupyter notebook entry point that demonstrates how to access the data used to fit our force fields. We are delighted to offer this alternative option and anticipate that it will provide another convenient method for you to leverage our work.

What is this Industry Benchmark Dataset and What Changed?
Our Industry Benchmarking dataset is a set of molecules designed to be highly representative of small-molecule drug discovery efforts. It is composed of molecules with less than 30−35 heavy atoms provided by our industry partners at BASF, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Janssen, Merck, KGaA, Roche, Genentech, Vertex, and XtalPi. [1] These molecules are expected to capture the research interests of our partners, while not being constrained by proprietary interest. Up to ten conformers were generated for each provided SMILES string and the geometries were minimized with Density Functional Theory geometry minimization at the B3LYP-D3BJ / DZVP level of theory.
Our new Industry Benchmarking v1.2 contains 9,835 unique molecules with 74,585 conformers. There were 29 conformers (from 7 unique molecules) removed from version 1.1 to create this dataset, due to unrealistic ring configurations in the initial geometry. These erroneous structures, identified by Alexandra McIsaac, persisted through a geometry optimization, resulting in unusually high energies. As a result, these structures are habitually removed from our internal pipelines. This new cleaned v1.2 is then a higher quality dataset for our stakeholders.
The contents of this dataset contains drug-like and biological scaffolds common in drug discovery efforts as shown in Figure 2. With 9,835 unique molecules, 3,760 can be categorized as one of the following: acridine, benzanilide, benzothiazole, beta-lactam, diphenylsulfone, flavone, imidazole, isoflavone, isoquinoline, nitroaromatic, phenoxybenzene, phenylsulfone, purine, quinoline, steroid-like, sulfonamides, tetralin, thiazole, or tropane. A molecule may be represented multiple times in this figure, where the identification of these substructures was achieved using the OpenEye Toolkit.
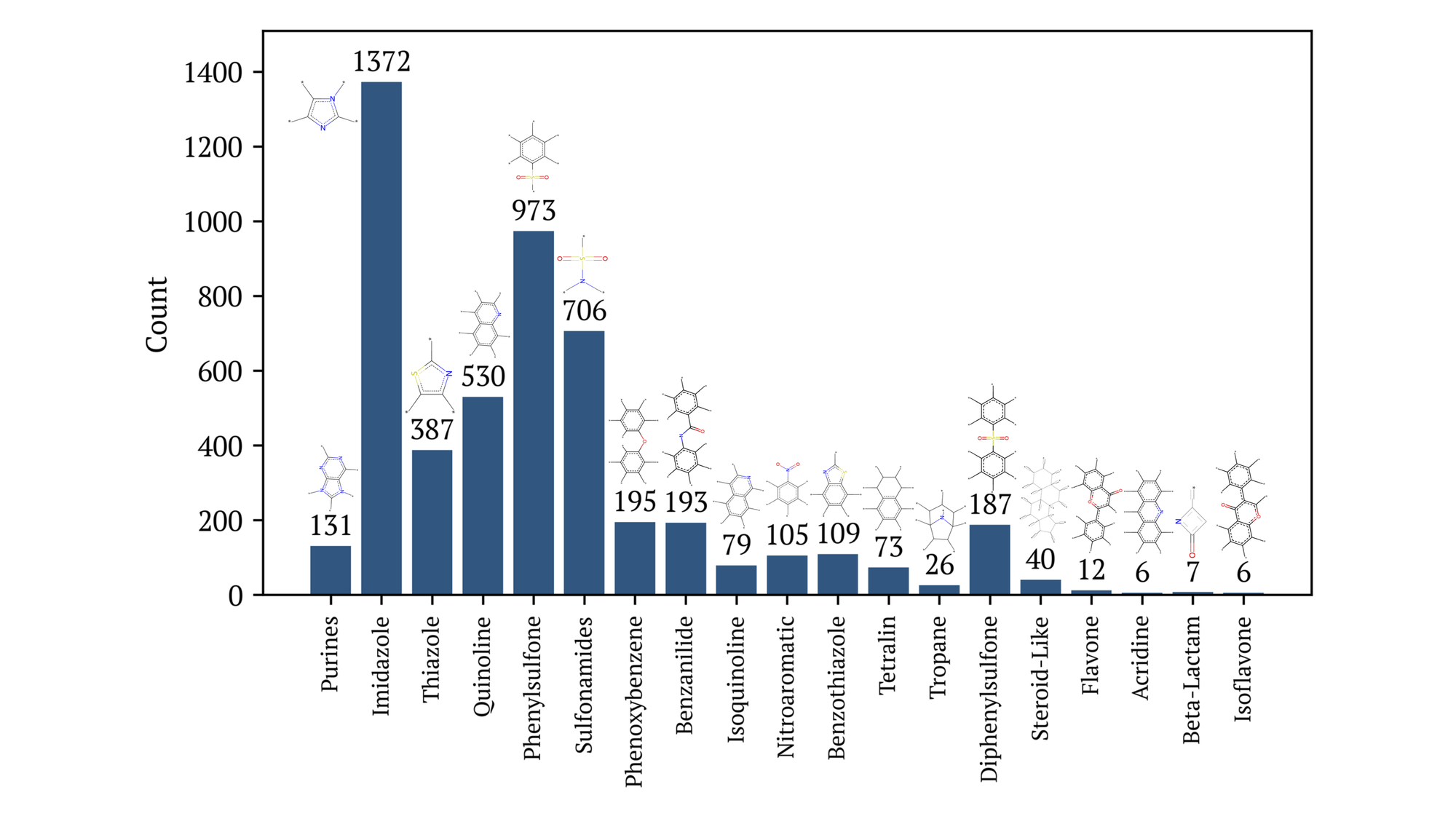
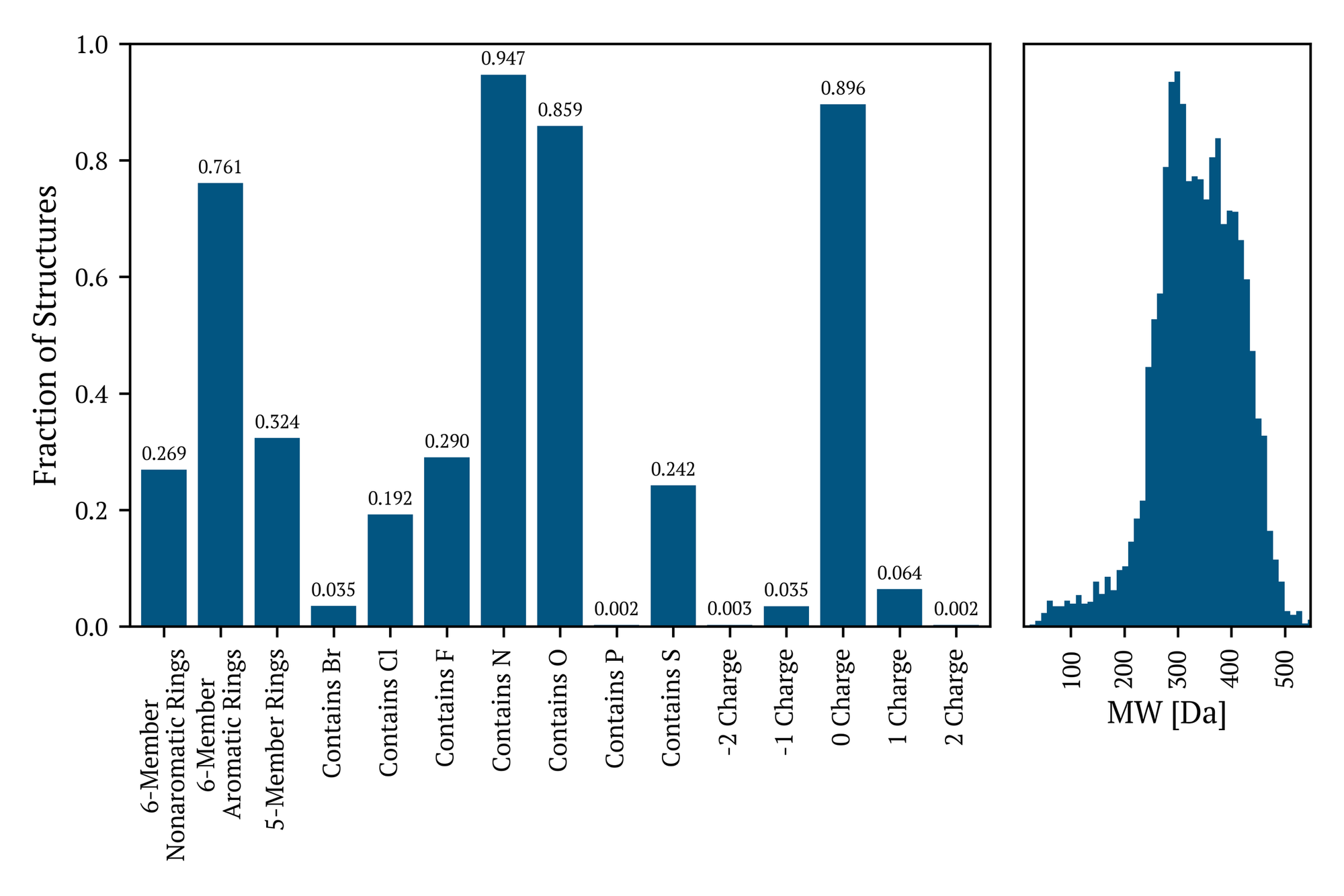
Although the remaining compounds do not possess moieties that precisely adhere to established drug-like scaffolds, they exhibit comparable structural motifs characterized by oxygen-, nitrogen-, and sulfur-containing hydrocarbon frameworks. Figure 3 illustrates the percentage of molecules in the full Industry Benchmark v1.2 that exhibit some element, ring type, or total molecular charge. These statistics are similar for the entire dataset and those remaining compounds that have not been identified as incorporating one of the assessed drug-like moieties. These molecules frequently incorporate single five- and six-membered ring systems (both aromatic and non-aromatic) and exhibit halogen substitution patterns. Each column in the left Figure 3 is adorned with the fraction of molecules in the Industry Benchmark v1.2 that contain these elements, moieties, and the distribution of total molecular charge. The right-hand side of Figure 3 presents the molecular weight distribution, with a minimum of 12 Da and a maximum of 1095 Da.
To conclude our evaluation of the Industry Benchmark v1.2 dataset, we employ CheckMol to identify functional groups through the application of our benchmarking tool, YAMMBS. Observe the disparity between the percentage of molecules with six-member aromatic rings depicted in Figure 3 and the significantly higher proportion of aromatic molecules identified in Figure 4. CheckMol detects all aromatic structures, encompassing fused rings that may be entirely or partially aromatic.
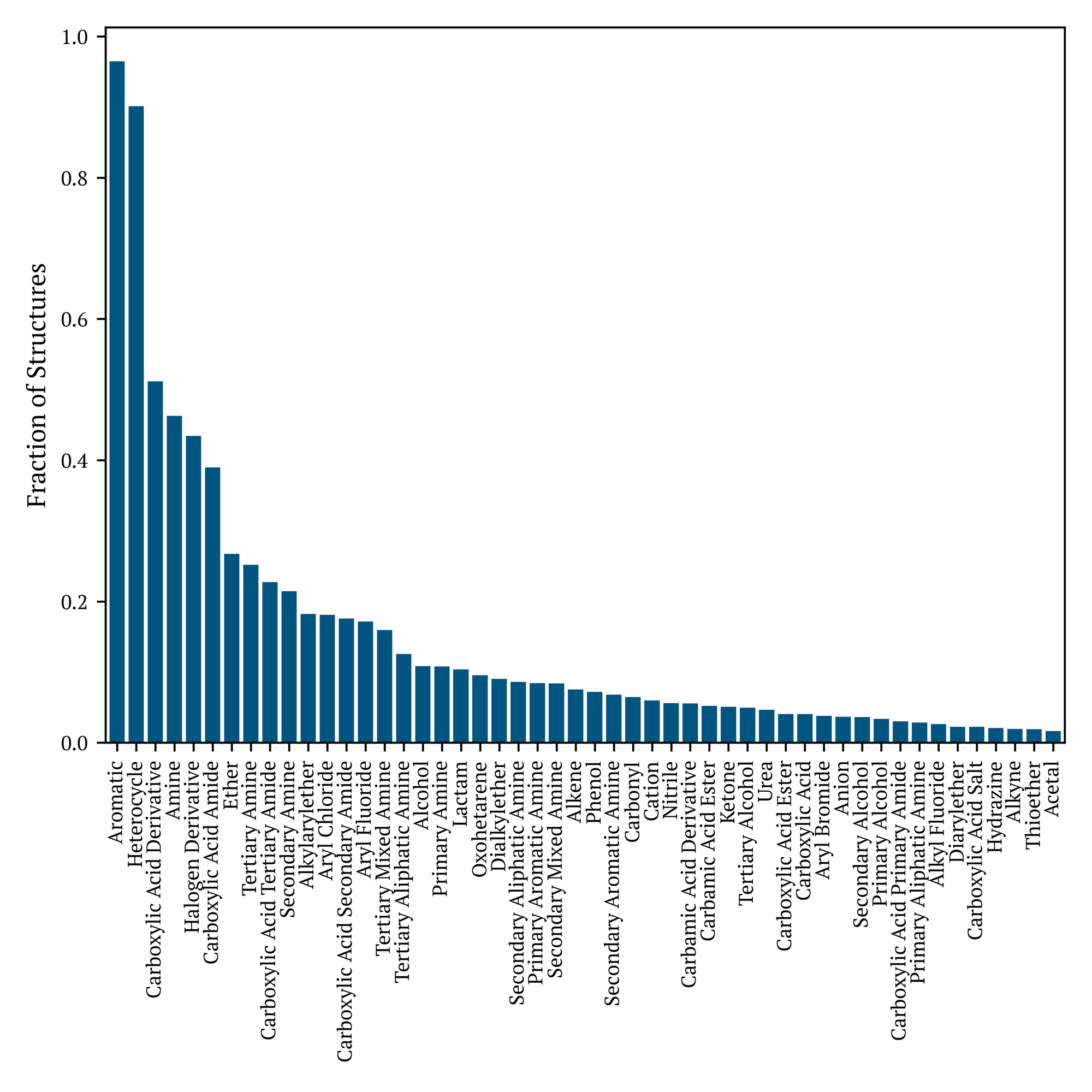
From the descriptions of the chemical space, it is evident that the Industry Benchmark v1.2 dataset comprises a diverse set of drug-like and drug-precursor molecules. While the dataset is largely neutral with an absolute charge of no more than two and little representation of phosphate among the structures, these molecules predominantly exhibit aromatic moieties incorporating nitrogen and, to a lesser extent, oxygen, with relatively low molecular weights. These structures contain a good portion of halogenated and sulfonated functional groups.
More About Open Force Field
Open Force Field is a nonprofit organization that develops a generalized forcefield for the scientific community and develops tools to create atomistic forcefields for molecular dynamics (MD) simulations. We firmly believe in providing our tools and data freely for transparent reproducibility and alternative use. If your organization wishes to collaborate with us, we are eager to contract with additional partners!
References
- D’Amore, L. et al. Collaborative Assessment of Molecular Geometries and Energies from the Open Force Field. J. Chem. Inf. Model. 62, 6094–6104 (2022).
- Wang, L. et al. The Open Force Field Initiative: Open Software and Open Science for Molecular Modeling. J. Phys. Chem. B 128, 7043–7067 (2024).
- Takaba, K. et al. Machine-learned molecular mechanics force fields from large-scale quantum chemical data. Chem. Sci. 15, 12861–12878 (2024).
- Boothroyd, S. et al. Development and Benchmarking of Open Force Field 2.0.0: The Sage Small Molecule Force Field. J. Chem. Theory Comput. 19, 3251–3275 (2023).
- Horton, J. T., Boothroyd, S., Behara, P. K., Mobley, D. L. & Cole, D. J. A transferable double exponential potential for condensed phase simulations of small molecules. Digit. Discov. 2, 1178–1187 (2023).
- Han, F. et al. Distribution of Bound Conformations in Conformational Ensembles for X-ray Ligands Predicted by the ANI-2X Machine Learning Potential. J. Chem. Inf. Model. 63, 6608–6618 (2023).
- Xue, B. et al. Development and Comprehensive Benchmark of a High-Quality AMBER-Consistent Small Molecule Force Field with Broad Chemical Space Coverage for Molecular Modeling and Free Energy Calculation. J. Chem. Theory Comput. 20, 799–818 (2024).
- Zheng, T. et al. Data-driven parametrization of molecular mechanics force fields for expansive chemical space coverage. Chem. Sci. 16, 2730–2740 (2025).
- Bonanni, D. et al. Condensation of Forcefield parameters from Machine Learning predicted statistical distributions for High-Throughput Virtual screening applications. Preprint at https://doi.org/10.26434/chemrxiv-2025-2tld1 (2025).
- Jiang, X. et al. Deep Residual Learning for Molecular Force Fields. Preprint at https://doi.org/10.26434/chemrxiv-2025-003lg-v3 (2025).
- Fusti-Molnar, L. QuantumFuture’s Conformational Database for Drug Discovery using Diverse Fragments of Chemical Interests from OpenFF Consortium | LinkedIn. LinkedIn Pulse https://www.linkedin.com/pulse/quantumfutures-conformational-database-drug-discovery-fusti-molnar-c1hlc/.
- geometry-benchmark-espaloma. Chodera Lab // Memorial Sloan Kettering Cancer Center (2023).
- Ries, B. & Hermann, M. TorsionProfiler. Boehringer Ingelheim (2025).